
CHAPS is a non-denaturing zwitterionic detergent for solubilizing membrane proteins and breaking protein-protein interactions. This detergent combines the useful properties of both sulfobetaine-type and the bile salt detergents. CHAPS is commonly used for protein solubilization in isoelectric focusing and two-dimensional electrophoresis, especially for non-denaturing (without urea) isoelectric focusing.
About CHAPS Detergent
This detergent has demonstrated excellent resolution of some sub-cellular preparations and plant proteins. Concentrations between 1-4% (v/v) are typically used in an isoelectric focusing gel. A commonly used isoelectric focusing sample solution consists of 8 M urea, 4% CHAPS, 50-100 mM dithiothreitol (DTT) and 40 mM Tris. Its small micellar molecular weight (6150) and high CMC (6-10 mM) allow it to be removed from samples by dialysis. It can be used as a non-toxic stabilizing agent for growth factors.
CAS: 75621-03-3
Chemical Formula: C32H58N2O7S
Synonyms: 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate; 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulphonate
Molecular Weight: 614.9 Da
Density: 1.01 g/mL at 20°C
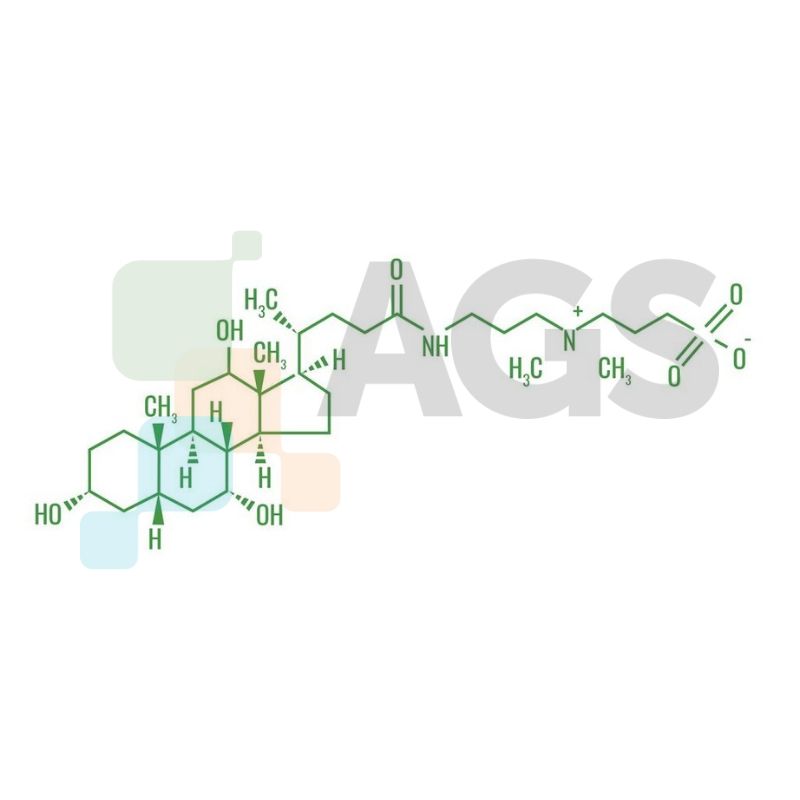
Chemical Structure of CHAPS:
Frequently Asked Questions
What is the physical appearance of CHAPS detergent?
This biodetergent is supplied as a white crystalline powder.
What is the critical micelle concentration (CMC) of CHAPS detergent?
The CMC value is 6-10 mM. Detergents with high CMC values are generally easy to remove by dilution; detergents with low CMC values are advantageous for separations on the basis of molecular weight. As a general rule, detergents should be used at their CMC and at a detergent-to-protein weight ratio of approximately ten.
In 0-0.1 M Na+, what is the aggregation number?
The aggregation number at this level is 4-14.
Is CHAPS detergent soluble in water?
Yes, this detergent becomes a clear and colorless to slightly-yellow solution in water. Solubility is 50 mg/mL at 20°C. It is preferable to avoid shaking and stirring mixtures when preparing CHAPS solutions.
How can I make a CHAPS 10% solution?
For a 10% solution (i.e., 1 g CHAPS solid into beaker followed by 9 g of water), cover beaker with watch glass and allow it to sit at room temperature for 30-60 minutes. Solutions of up to 1 M (60%) can be made in this way.
What is the stability of this detergent?
CHAPS biodetergent is moisture sensitive and hygroscopic.
Can CHAPS detergent be removed from samples by dialysis?
Yes, this detergent’s small micellar molecular weight and high critical micelle concentration (6-10 mM) allow it to be removed from samples by dialysis.
What is the difference between CHAPS and CHAPSO detergents?
A related detergent, called CHAPSO, has the same basic chemical structure with an additional hydroxyl functional group. Both detergents have low light absorbance in the ultraviolet region of the electromagnetic spectrum, which is useful for laboratory workers monitoring ongoing chemical reactions or protein-protein binding with UV/Vis spectroscopy.
What concentration of CHAPS is used for preparing IEF gel?
Concentrations between 2-4 % are typically used in an IEF gel. CHAPS is commonly used for non-denaturing IEF (without urea) and has been shown to give excellent resolution of some subcellular preparations and plant proteins.
What is the difference between CHAPS and Triton X100?
Triton X100 is non-ionic. It has both hydrophilic and hydrophobic regions, but no net charges. CHAPS detergent is zwitterionic. It has hydrophobic regions but also a head group with a negative charge (in normal saline). In addition, Triton X100 forms large (greater than 90,000 MW) aggregates when concentration rises above 0.25 mM. CHAPS on the other hand forms smaller aggregates (6,000 MW) when concentration rises above 10 mM.
CHAPS Buffer Preparation Protocol
CHAPS detergent can be used to lyse cells under non-denaturing conditions and is recommended for the preparation of cytoplasmic cell lists to be used with our caspase signaling pathway antibodies. Buffer should be prepared immediately before use.
Components
- 10x CHAPS detergent (5 mL)
- 1x concentration: 50 mM PIPES/HCl (pH 6.5), 2 mM EDTA, 0.1% CHAPS, 20 ug/mL Leupeptin, 10 ug/mL Pepstatin A, 10 ug/mL Aprotinin
- 1x concentration: 5 mM DTT
Buffer Preparation Protocol
- Immediately before dilution, invert the 10X CHAPS several times to suspend all buffer components.
- Add 1/10 volume of 10X CHAPS to 9/10 volume of Milli-Q water or equivalently purified water.
- Add DTT to final concentration of 5 mM (1:200) dilution and PMSF to final concentration of 1 mM.
Sample Preparation using CHAPS Detergent
- Treat cells by adding fresh media containing regulator for desired time.
- Aspirate media from cultures, wash cells three times with PBS; aspirate. Scrape cells into PBS and spin down to pellet.
- Lyse cells by adding ice-cold 1X CHAPS (1 volume of cell pellet). Resuspend cells in the buffer, freeze at -80°C and thaw twice, centrifuge the lysate cold at 14,000 rpm. Keep the supernatant and discard the pelleted cell debris.
- Add SDS sample buffer and heat sample to 95-100°C for 5 minutes. Cool on ice.
- Microcentrifuge for 5 minutes.
- Load 5 uL onto SDS-PAGE gel (10 cm x 10 cm)
Storage
Recommended storage temprature: -20°C
Cell Lysis and Immunoprecipitation with CHAPS Detergent
The CHAPS immunoprecipitation buffer (CIB-1) is formulated to maintain intermolecular interactions following transmembrane and cytosolic protein extraction. The CHAPS buffer is ideal for pull down co-immunoprecipitation (i.e.Co-IP) analysis, since it maintains protein conformation as well as protein complex assembly. Following cell lysis and cellular protein solubilization into CHAPS buffer, assembled proteins can be isolated together using immunoprecipitation technique. An immunoprecipitating antibody is added to bind to one of the assembled protein targets.
A solid support, such a Protein-G Sepharose, is then added to non-covalently link and bind to the antibody-protein complex and make it insoluble. Following low-speed centrifugation, sedimentation and wash, the solid support-antibody-protein complex is isolated together. Proteins assembled with the protein targeted by the immunoprecipitating antibody become co-immunoprecipitated. By developing Western blots with a different antibody targeting a co-assembled protein, the researcher can characterize and quantify the interaction among two or more protein species. Functional biochemical activity assays can also be employed to characterize co-immunoprecipitated and assembled biomolecules.
Components
- Microcentrifuge at 4°C
- Rocker table
- Cold room or refrigerated case
- Antibody suitable for immunoprecipitation as specified by the vendor. Generally, the immunoprecipitating antibody must recognize the NATIVE conformation.
- Western blots applications: Antibody suitable for Western blot detection as specified by the vendor. This antibody must recognize the DENATURED state of the targeted protein.
Suggested Controls and Samples for Comparisons
- Perform immunoprecipitation protocol with an irrelevant antibody to control for appearance of spurious protein bands.
- Resolve in Western blots the original unfractionated cell lysate, the unbound fraction (e.g. the fraction containing proteins not bound to the protein-A or G solid support) and the immunoprecipitated materials to confirm fractionation and enrichment.
- Develop a Western blot that detects the protein directly targeted by the immunoprecipitating antibody. This procedure confirms that immunoprecipitation occurred.
A. Protocol for Cell Culture
It is recommended that cells are cultured to 80-90% confluency prior to performing cell lysis and immunoprecipitation. Cells should be washed free of serum proteins using PBP prior to performing immunoprecipitation to prevent appearance of non-specific serum protein bands in downstream Western blots.
B. Protocol for Cell Lysis
We recommend using 300 uL of CHAPS solution for one to three 10 cm cell culture dishes of lysed cells. Scale accordingly for other numbers or sizes of cell culture dishes according to the surface area of the dish. Prior to lysis, make the CHAPS Buffer ice cold and add protease and phosphatase inhibitors.
Cells should be washed free of serum proteins using PBS prior to performing immunoprecipitation to prevent appearance of non-specific serum protein bands in downstream Western blots. Remove the cell culture medium and gently wash the 10cm cell culture dish with 5 mL PBS, three times. Use a gentle stream of PBS to prevent excessive loss of cells from the plate. After the washes, cover the cell culture dish with the lid and place the cell culture dish on a bed of ice.
- Lyse cells and generate a supernatant fraction rapidly as follows: Dispense 300 uL ice cold CHAPS with protease/phosphatase inhibitors over the cell layer, rotate the plate by hand to cover cells with a film of CHAPS, then immediately dislodge the cells with a cell scraper. Use a transfer pipette with a wide opening to siphon the cells into a 1.5 mL microcentrifuge tube. The 300 uL Chaps solution can also be transferred sequentially to up to three separate 10 cm plates of cells before the cell suspension is placed into a 1.5 mL microcentrifuge tube.
- Place the 1.5 mL microcentrifuge tube with cell suspension on ice for 10 min: Strongly tap the tube several times during this 10 min period to facilitate cell membrane dissolution. You can also use a rocker table to rotate the cell suspension to further facilitate cell membrane dissolution. Do not vortex the cell lysate if immunoprecipitation is planned.
- Centrifuge the cell lysate in a cooled microcentrifuge at full speed for 15 min to partition supernatant and pellet. Collect the supernatant fraction, which contains extracted membrane and cytosolic proteins, and dispense this supernatant into another 1.5 mL microcentrifuge tube that is placed in ice. This transferred supernatant corresponds to the unfractionated supernatant fraction. Set aside a small aliquot for comparisons to the immunoprecipitated materials that are generated later in the protocol. The unfractionated supernatant can be stored at -20°C, or -80°C for longer term storage.
C. Protocol for Immunoprecipitation
- Add immunoprecipitating antibody to the unfractionated supernatant fraction, using the antibody titer recommended by the manufacturer. Place the tube with the immunoprecipitation reaction on a rocker table under refrigeration (such as a cold room, or refrigerated case) for 15-30 min to mix the antibody-supernatant mixture. You may opt to initially try the shorter 15 min period for immunoprecipitation, a time period that is more apt to maintain low affinity interactions.
- Directly dispense 60 uL of immunoprecipitation beads (e.g. Sepharose-G beads, or protein-A beads) into every 300 uL to 2 mL aliquot of unfractionated supernatant with immunoprecipitating antibody. Place this tube on a rocker table for another 30 min to 1 hr under refrigeration to generate an insoluble solid support–antibody-protein complex.
- Use a refrigerated microcentrifuge at 3000 rpm for 5 minutes to sediment the immunoprecipitation beads. The resulting supernatant is the unbound fraction. Collect the unbound fraction without disturbing the immunoprecipitating bead pellet. Store the unbound fraction at -20°C or -80°C.
- Add an additional aliquot of fresh 300 uL ice cold CHAPS with protease and phosphatase inhibitors to the immunoprecipitation beads. Gently rotate the tube 180° by hand three times and centrifuge again at 3000 rpm for 5 minutes. Remove and discard the wash supernatant. Repeat this wash procedure two more times. After the last wash, use a microcentrifuge to sediment the immunoprecipitation beads at 14000 rpm for 15 min. Remove as much of the wash solution as possible using a pointed plastic Pasteur pipette. The immunoprecipitated fraction, which is bound to the beads, becomes sedimented at the bottom of the tube.
D. Protocol for Elution of the Immunoprecipitated Fraction from Beads and Western Blot
- There are several methods to elute the immunoprecipitated proteins from the solid support to release a soluble immunoprecipitated fraction. The simplest method applicable for subsequent Western blotting is to apply Laemmli Sample Buffer (LSB; with mercaptoethanol) directly to the immunopreciptation beads: Add 100l LSB to each 60 uL of sedimented immunoprecipitation beads. Vortex the LSB – immunoprecipitate solution at full speed for 30 sec, and then heat at 60°C for 10 min. Now sediment the beads using low speed centrifugation (3000 rpm) for 5 min. Collect the supernatant; The immunprecipitated proteins (along with the immunoprecipitating antibody) will be released into the supernatant.
- The supernatant can be resolved in subsequent Western blots. Alternatively, if your protein of interest aggregates easily when heated at high temperature in LSB, heat instead at 37°C for 30 min. and follow the same aforementioned steps. Resolve in consecutive gel lanes and Western blots, 1) the unfractionated fraction, 2) unbound fraction and 3) immunoprecipitated fraction. With successful immunoprecipitation, you should observe enrichment of your protein of interest, along with any co-assembled proteins in the immunopreciptated fraction.
E. Protocol for Tissue Homogenization Prior to Immunoprecipitation
This process should be peformed on ice.
- Pulverize approximately 90 uL of tissue. Place tissue in a 1.5 mL round bottom microcentrifuge tube.
- Add general phosphatase and protease inhibitor cocktails to 500 uL of ice-cold CHAPS Lysis and Immunoprecipitation Buffer
- Add 500 uL CHAPS buffer with inhibitors to pulverized tissue.
- Homogenize tissue with a mini pestle-homogenizer using 15 strokes, 3 seconds/stroke on ice.
- Centrifuge 12,000 g for 15 min at 4°C
- Remove supernatant (without lipid layer) and transfer into another 1.5 mL tube
- Centrifuge again at 12,000 g for 15 min at 4°C
- Transfer supernatant to another tube. The supernatant fraction contains the extracted proteins that can be used for immunoprecipitation.
Troubleshooting
- Immunoprecipitation did not occur. Resolution: 1) You may have to empirically identify an antibody for immunoprecipitation that recognizes the native conformation of the epitope. 2) The protein complex is not maintained outside of a live-intact cell. Cross-linking procedures using membrane permeable cross-linking reagents and live cells may be required to capture the interaction and assembly among proteins.
- The IgG heavy chain of the immunoprecipitating antibody (that was dispensed in the immunoprecipitation solution) migrates in gels at the same position as a protein of interest, and therefore masks its appearance in a Western blot. Resolution: For Western blot development, use an antibody derived from an animal host different from the immunoprecipitating antibody. In this case, the appropriate secondary-HRP conjugated antibody will not bind appreciably to the immunoprecipitating antibody that was transferred to the Western blot, preventing the appearance of an overlaying Western blot band.